










 发布时间:2024/01/12 点击数:
发布时间:2024/01/12 点击数:本篇将介绍利用肠-肝微流控器官芯片系统提高口服生物利用度的预测。

本文概况
本研究介绍了CN-Bio的肠肝微流控器官芯片系统(OOC&MPS),使用数学建模方法优化实验设计,从16种不同的化合物选择了4种药物进行口服生物利用度实验。与动物模型相比,MPS预测人体口服生物利用度的准确性得到了改善。这一研究为更具人体相关性的体外方法在药物研发中的应用提供了潜在机会,有望提高药物开发的成功率。
引言
药物的开发过程中,需要理解药物动力学(PK)特性,其中之一便是生物利用度,其定义为药物在肠道吸收和肝脏首过代谢后进入体循环的比例。对于生物利用度较低的药物,往往难以保持在治疗窗口内。通常需要使用更大剂量才能达到治疗效果,但这样做有诱发不良安全效应的风险,需要对生物利用度进行估计,以指导药物开发过程,因此其准确性与临床试验的成功或失败有较大的相关性。
口服生物利用度通常使用动物(如狗、大鼠、小鼠和非人类灵长类动物)进行估计,其中狗、大鼠、小鼠和非人类灵长类动物是较常用的。然而,这些物种与人类生物利用度的总体相关性(R2 = 0.34)较弱,正如 Musther 等人的一项重要研究所述,他们调查了184种化合物的生物利用度1。
此外,与动物研究相关的较高成本和伦理考虑引发了以下问题:是否存在更具人体相关性的体外方法,以获得更准确的口服生物利用度估计?
微生理系统(MPS),通常被称为“芯片上的器官”(organ-on-a-chip),已成为在临床前药物ADME研究中获得相关人体数据的潜力工具。它们旨在通过在3D支架中培养细胞,并在灌流条件下模拟血液流动,来复制人体组织的结构和功能特性。到目前为止,MPS的发展主要集中在单器官组织模型上,但更复杂的多器官系统正在出现,以满足改善体外到体内PK预测的需求。对于口服生物利用度,需要结合肠肝MPS来模拟药物在肠道中的吸收和随后在肝脏中的首过代谢过程。
研究目标
在这里,我们描述了一种使用PhysioMimix OOC MPS-TL6消耗性培养板来估计人体口服生物利用度的肠-肝MPS。该培养板与CN Bio的PhysioMimix多器官芯片系统设备兼容,由六个独立孔组成,每个独立孔内有两个腔室,分别模拟肠道和肝脏。培养基通过连接通道在各腔室之间循环。肠道MPS由上皮和腺细胞系的混合物组成,形成了Transwell膜内侧的屏障。肝脏MPS由原代人肝细胞(PHH)组成,在多孔胶原蛋白涂层支架上形成3D微组织。
通过使用单个MPS-TL6培养板模拟口服和静脉注射(IV)剂量给药,进行口服生物利用度的估计。采用数学模型来协助设计和优化实验方法。
材料与方法
冷冻保存的可培养性原代人肝细胞(PHH)由ThermoFisher购得。每个 MPS-TL6培养板(CN Bio Innovations Ltd)的肝脏隔室中接种了0.6 x 106 PHH,并在含有500 nM羟化可的松(Sigma)的肝细胞维持培养基中进行培养。肠道屏障是由Caco-2/HT-29细胞(ReadyCell)的混合物组成,在肝脏培养(PHH)的第5天之前,在含有血清的肠道培养基中预培养了21天,然后添加到MPS-TL6板中。以下药物的口服(100µM无血清肠道培养基)和静脉注射(10 µM肝细胞维持培养基)剂量被制备:阿司匹洛、苯妥英、萘普生和甲基泼尼松。
对于每种药物,使用一个MPS-TL6培养板来估计生物利用度。六个独立孔中的三个用于模拟口服给药方案,剩下的三个用于静脉注射给药方案。在第5天,将肝细胞维持培养基(不含药物)加入口服剂量孔的肝脏和肠道基底腔室。对于静脉注射孔,药物被注入肝脏和肠道基底腔室。在更换培养基后,肠道MPS Transwell随后插入MPS-TL6培养板。对于口服剂量孔,药物被加入Transwell的顶部。对于静脉注射孔,无血清肠道培养基(不含药物)被加入Transwell的顶部。
在0、1、4、6、24和48小时时采集培养基样本,并通过液相色谱-质谱联用(LC-MS)分析来确定肝脏隔室中母药的浓度。使用GraphPad Prism来估算口服和静脉注射浓度曲线下面积(AUC)。然后,使用以下计算方法估算口服生物利用度:
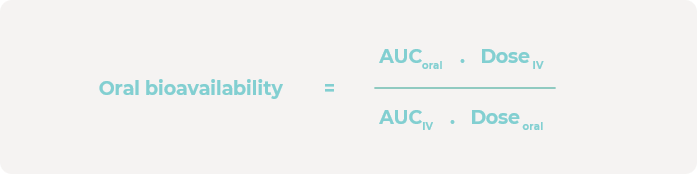
在第4天和第7天使用P450-Glo CYP3A4酶活性测定(Promega)评估PHH的代谢潜能。通过测量跨膜电阻(TEER)来评估药物给药实验前后的肠道屏障稳定性。作为对照,还在24孔培养板中以相同的顶部(无血清肠道培养基)和底部(肝细胞维持培养基)培养基,静态培养肠道Transwell,同样进行了TEER评估,以与肠肝实验进行比较。
药物的选择用于通过肠肝MPS估计生物利用度,源自Musther等人的一项关于人体和动物生物利用度比较的研究1。选择范围缩小到那些在Transwell上的Caco-2屏障中具有肝细胞清除率和渗透性参考数据的药物2,3。数学建模需要这两个输入参数,我们用它来指导和优化实验设计。选择了16种具有生物制药药物分布分类系统(BDDCS)1或2类的化合物,其主要代谢途径是肝脏,用于数学建模。使用R编程语言进行了数学建模,使用常微分方程(ODE)来描述MPS-TL6培养板中随时间的浓度变化。包RxODE用于在R中模拟ODE4。在这16种药物中,LogD值小于3的药物:阿司匹洛、苯妥英、萘普生和甲基泼尼松,被选中用于通过肠肝MPS估计生物利用度。LogD值大于3可能会具有不良的理化药物特性,如溶解度差和蛋白结合增加,使得估计PK特性更具挑战性。
使用Maier等人描述的模型5,估算了16种药物在小肠中的浓度。简而言之,该模型基于对泊沙康唑的体内研究,该研究在酸性或中性溶液中将40毫克(57µM)的药物输送到志愿者的胃中6。十二指肠中的至高浓度分别达到26.3 ± 10.3µM或13.6 ± 5.8µM,相当于将药物溶解在300毫升水中,吸收率为90%6,7。我们使用这种关系来估算药物在吸收前后的小肠中的浓度,根据英国国家药典(BNF)、世界卫生组织解剖治疗化学分类(ATC)或医学专业月刊(MIMS)数据库中的16种药物的临床推荐剂量。
结果与讨论
1、MPS-TL6中口服生物利用度的估算
使用PhysioMimix 多器官系统(图1A),我们展示了多器官肠肝MPS在体外估算口服生物利用度的应用。在这项研究中,每个MPS-TL6培养板的井被分为两个给药方案,以估算口服和静脉注射剂量后的药物生物利用度(图1B)。对于口服给药,药物被加入到肠道MPS的顶部,而对于静脉注射,药物被加入到循环于肠道和肝脏隔室之间的介质中。在第5天给药,在原代肝细胞接种和将肠培养物插入MPS-TL6培养板后,检测窗口为48小时(图1C)。
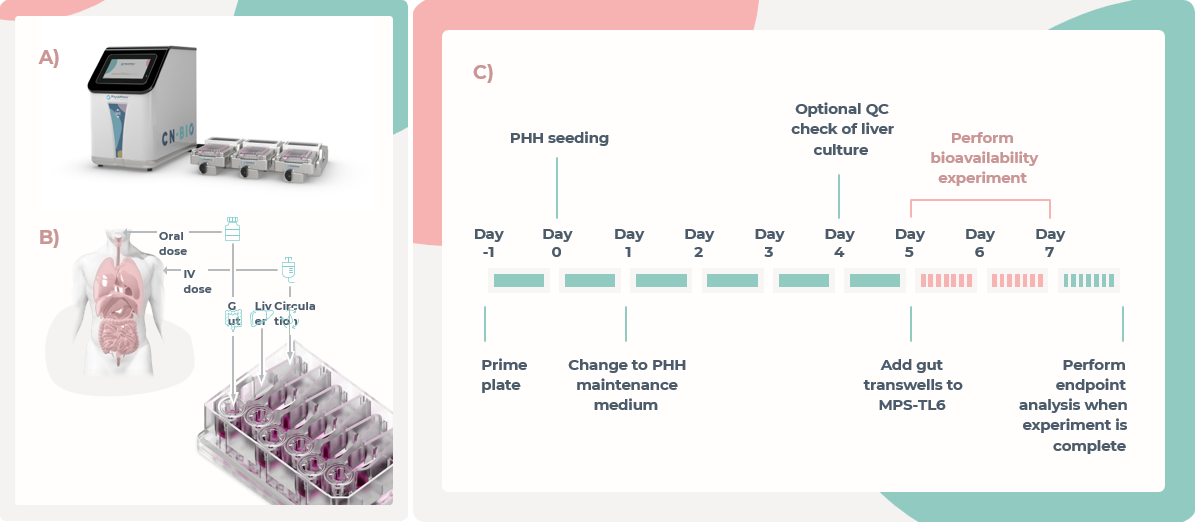
图1.MPS-TL6中口服生物利用度的估算
A) MPS-TL6中的流体由PhysioMimix多器官系统控制流动。
B) 为了估计口服生物利用度,药物被添加为口服剂量,进入肠腔室,或作为静脉注射剂量,流经MPS-TL6板的肝脏和肠隔室之间的循环介质。
C) 生物利用度估计的实验时间线。PHH被种植到多孔支架中,在肝隔室内形成3D微组织,第1天更换培养基。在第4天,采集介质样本,并评估QC指标,以确保功能稳定。在第5天,预培养的肠道Transwell被添加到MPS-TL6板的肠隔室中。随后通过流体流动将肠和肝隔室连接在一起,然后进行药物给药。在48小时内采集样本,以获得药物浓度曲线。
2、口服生物利用度实验的设计与优化
利用数学建模方法,通过描述MPS-TL6培养板每个隔室中化合物浓度的方程(图2A),来协助指导生物利用度实验的设计。从一项研究中选择了16种化合物(R2 = 0.32,图2B)进行数学建模,该研究调查了184种化合物的动物与人体生物利用度(R2 = 0.34),发现相关性较弱1。数学建模被用于优化多个系统参数,旨在设计一个接近人体生物利用度值的实验。例如,在MPS-TL6中评估了一系列隔室容积和流速,这些值介于板允许的至小值和至大值之间,还研究了样本点的时间和频率,以阿司匹洛为例(图2C)。模型发现,将研究时间限制在仅6小时,或者在早期时间点进行过少的采样,将导致生物利用度的估计不准确。
对于口服药物给药,我们旨在使用相同的浓度,且该浓度与小肠中的有效药物浓度相关,相关的浓度会影响避免药物饱和肠道MPS并可能诱发毒性并导致屏障丧失。使用Maier等人描述的方法,基于药物的临床推荐剂量和小肠中的静息水容积,来估算药物在肠道的浓度5。我们估计了16种药物在小肠中的中位数药物浓度,在吸收前和吸收后分别为659.3 µM和97.2 µM(图2D)。为了降低药物引起肠道毒性的可能性,我们选择了一个相关的口服药物浓度,即100µM,位于估算的较低端,用于所有生物利用度实验。为了测试我们的肠肝MPS模型准确预测口服生物利用度的能力,从16种药物中选择了LogD值小于3的4种药物:阿司匹洛、苯妥英、萘普生和甲基泼尼松。

图2.口服生物利用度实验的设计与优化
A) 描述MPS-TL6培养板中每个隔室化合物浓度的数学模型示意图。对16种化合物的口服生物利用度进行了建模。
B) 动物模型对这16种化合物的生物利用度预测较弱1。
C) 运行数学模型以优化肠肝MPS实验参数,包括更佳采样时间和采样频率。以阿司匹洛为例展示,旨在尽可能接近其已知的人体生物利用度值。
D) 使用Maier等人描述的方法估计16种化合物在吸收前后的小肠中浓度,该方法考虑了每种药物的临床剂量和小肠中的静息体积5。
3、在MPS-TL6培养板中,肝脏和肠道功能的质量控制指标得以维持
生物利用度估计期间保持了PHH的代谢能力和肠道屏障的完整性等质量控制指标(图3)。这表明生物利用度估计是基于功能稳定的体外多器官肠肝MPS得出的,因此我们对其准确性具有信心。
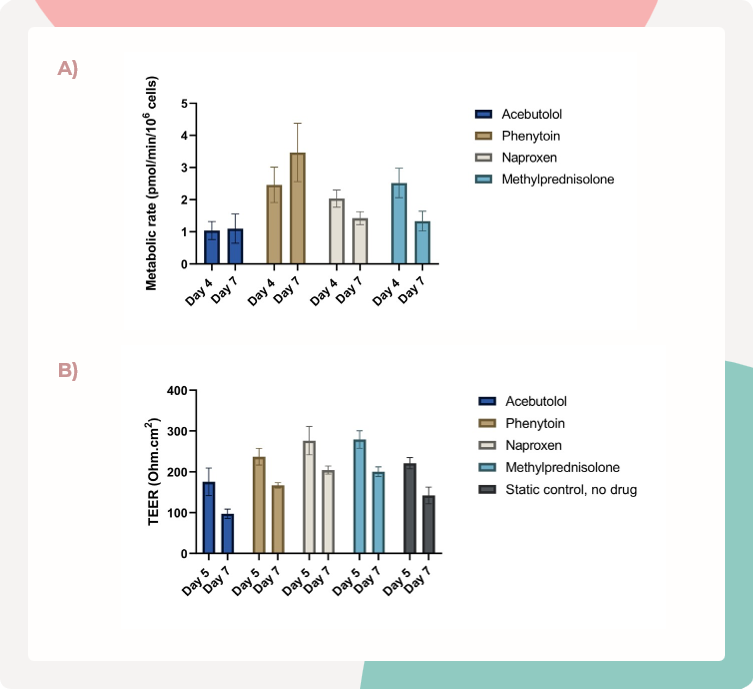
图3.在MPS-TL6培养板中,肝脏和肠道功能的质量控制指标得以维持
选择了四种化合物进行口服生物利用度实验:阿司匹洛、苯妥英、萘普生和甲基泼尼松。
A) 通过P450-Glo CYP3A4酶活性测定对肝脏MPS的代谢能力进行了评估,并在整个研究过程中得以维持。
B) 肠道MPS中的上皮屏障稳定性得以维持,通过在将肠道Transwell添加到MPS-TL6(第5天)和研究结束时(第7天)对TEER进行测量来进行评估。
4、与动物模型相比,MPS-TL6与人体生物利用度的相关性得到了改善
在MPS-TL6培养板中,对这4种药物的口服和静脉给药进行了比较,通过肠道屏障的渗透性和肝脏细胞的清除率,这些是浓度曲线的主要驱动因素(图4)。观察到的药物清除程度与报告的体外非结合内源性清除率2(CLint,u,图4A)相符。对于阿司匹洛、苯妥英和甲基泼尼松,肠肝MPS对人体生物利用度的预测优于动物模型(图4B)。对于萘普生,动物模型准确预测生物利用度,而我们的肠肝MPS表现相似(图4B)。
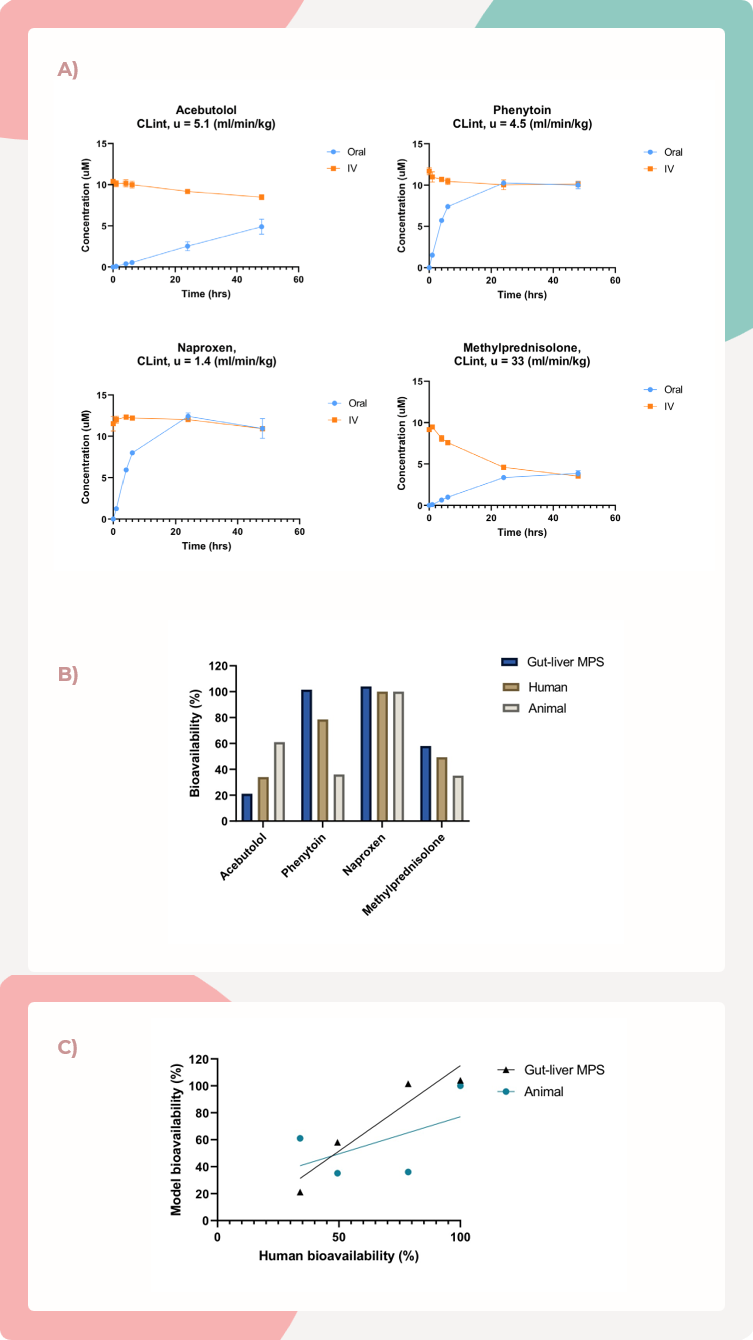
图4. 与动物模型相比,MPS-TL6与人体生物利用度的相关性得到了改善
A) 阿司匹洛、苯妥英、萘普生和甲基泼尼松在肝隔室中进行口服和静脉注射途径的浓度随时间变化图。每种化合物的清除程度与报告的体外非结合内源性清除率(CLint, u)相符。
B) 肠肝MPS显示对阿司匹洛、苯妥英和甲基泼尼松的人体生物利用度预测有所改善,而萘普生与动物模型的准确预测相当。
C) 与使用动物获得的值相比,对这四种化合物进行实验测试后,与人体生物利用度的整体相关性得到了改善。
结论
使用具有较高生物利用度值的药物,肠-肝微生理系统(MPS)比动物模型能更准确地预测人体生物利用度。对于4种测试的化合物中的3种,肠-肝MPS的预测效果更好,对于第4种,肠-肝MPS的预测效果与动物模型相似。与动物实验相关的伦理和成本考虑相比,这种体外方法可以在不到10天的时间内,仅使用单个MPS-TL6板来预测每种药物的人体生物利用度。肠-肝MPS有潜力在药物开发的早期提供与人体相关的生物利用度进行预测估计,有助于确保药物在临床试验中的成功或防止失败。
参考文献
1.Musther, H., Olivares-Morales, A., Hatley, O. J. D., Liu, B. & Hodjegan, A. R. Animal versus human oral drug bioavailability: Do they correlate? Eur. J. Pharm. Sci. 57, 280 (2014).
2.Hallifax, D., Foster, J. A. & Houston, J. B. Prediction of Human Metabolic Clearance from In Vitro Systems: Retrospective Analysis and Prospective View. Pharm. Res. 2010 2710 27, 2150–2161 (2010).
3.Li, C., Liu, T., Cui, X., Uss, A. S. & Cheng, K.-C. Development of In Vitro Pharmacokinetic Screens Using Caco-2, Human Hepatocyte, and Caco-2/Human Hepatocyte Hybrid Systems for the Prediction of Oral Bioavailability in Humans: http://dx.doi.org/10.1177/1087057107308892 12, 1084–1091 (2016).
4.W, W., KM, H. & DA, J. A Tutorial on RxODE: Simulating Differential Equation Pharmacometric Models in R. CPT pharmacometrics Syst. Pharmacol. 5, 3–10 (2016).
5.Maier, L. et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 555, 623 (2018).
6.Hens, B., Brouwers, J., Corsetti, M. & Augustijns, P. Supersaturation and Precipitation of Posaconazole Upon Entry in the Upper Small Intestine in Humans. J. Pharm. Sci. 105, 2677–2684 (2016).
7.Mudie, D. M. et al. Quantification of Gastrointestinal Liquid Volumes and Distribution Following a 240 mL Dose of Water in the Fasted State. Mol. Pharm. 11, 3039–3047 (2014).

扫一扫